

67 гостей
спадкові хвороби
- § 54. Етіологія спадкових хвороб
- § 55. Спадкові хвороби, обумовлені генними мутаціями
- § 56. Хвороби, успадковані за домінантним типом
- § 57. Хвороби, успадковані за рецесивним типом
- § 58. Спадкові хвороби, зчеплені з підлогою
- § 59. Хромосомні хвороби
- § 60. Патогенез спадкових хвороб
- § 61. Поширеність спадкових хвороб і соціальні аспекти генетики людини
Частина перша. Загальна нозологія
Розділ IV. РОЛЬ спадковості, КОНСТИТУЦІЇ ТА ВІКУ В ПАТОЛОГІЇ
Глава 1. Спадкові хвороби
Спадкові фактори відіграють істотну роль в патології людини. Вони визначають виникнення так званих спадкових захворювань, а також впливають на розвиток хвороби і механізми одужання при багатьох неспадкових формах патології.
Розвиток патологічних процесів залежить від взаємодії генотипу і умов зовнішнього середовища. Під генотипом розуміється сукупність спадкових факторів (генів) організму, що детермінують його спадкові властивості. До спадкових захворювань належать хвороби, що розвиваються в результаті патологічних змін генотипу.
У розвитку спадкових хвороб має значення зовнішнє середовище. Умови зовнішнього середовища можуть сприяти, а можуть і перешкоджати прояву патологічних спадкових ознак. При деяких захворюваннях, наприклад гемофілії, хондродистрофії, дальтонізмі, атаксії мозочка, розвиток хвороби визначається патологічним геном, т. Е. Залежить від ендогенних факторів. У цих випадках патологічний ген має високу пенетрантністю *, близькою до 100%. (* Пенетрантность - виражене у відсотках відношення числа особин, які проявляють патологічний ознака, до числа володарів відповідного гена.) Це спадково обумовлені захворювання.
В інших випадках (подагра, цукровий діабет, атеросклероз) виникнення хвороби залежить від певного взаємодії генетичних факторів і факторів зовнішнього середовища: пенетрантность гена коливається і в значній мірі схильна до впливом середовища. Це хвороби зі спадковою схильністю.
Нахил до спадкових хвороб може проявитися у вигляді зміни нормальної реакції на певні зовнішні впливи. Наприклад, при нахилі до цукрового діабету виявляється зміна нормальної реакції на цукор (патологічні цукрові криві).
Спадкові хвороби можуть проявлятися в різному віці. Деякі з них виявляються ще в ембріональному періоді розвитку і у новонароджених, наприклад гемофілія, спадкова форма глухонімоти, іхтіоз ( "риб'яча луска"). Інші захворювання виникають в дитинстві, а деякі виявляються клінічно вперше в зрілому або навіть похилому віці. Так, подагра розвивається після 40-45 років, хорея Геттінгтона - після 35 і навіть 60 років.
Для оцінки відносної ролі факторів зовнішнього середовища і генетичних факторів у виникненні спадкового захворювання користуються поняттям конкордантности близнюків. Близнюки називаються конкордантность, якщо вони однакові по досліджуваній ознаці. Так, при наявності у обох близнюків одного і того ж захворювання або при відсутності у обох цього захворювання говорять про конкордантности. Якщо ж близнюк захворює, а інший ні, пару називають дискордантній (табл.10).
Таблиця 10. Частота захворювання другого близнюка (у відсотках) в разі захворювання першого)Спадкові та інфекційні хворобиБлизнюкимонозиготнідизиготні
Заяча губа 33 5 Маніакально-депресивний психоз 96 19 Вроджена звуження воротаря 67 3 Пневмонія 58 43 Клишоногість 32 3 Дифтерія 50 38 Цукровий діабет 65 18 Свинка 82 74 Шизофренія 69 10 Кашлюк 97 93 Кір 98 94
З таблиці видно, що інфекційні хвороби однаково часто вражають як дизиготних, так і спадково ідентичних однояйцевих близнюків. При спадкових захворюваннях конкордантность у монозиготних близнюків значно вище, ніж у дизиготних.
§ 54. Етіологія спадкових хвороб
Причиною спадкових хвороб є патологічні мутації, т. Е. Раптові стрибкоподібні зміни спадкових властивостей організму. Мутації виникають в результаті дії на організм і його спадковий апарат факторів зовнішнього середовища. Мутагенну дію мають різні види іонізуючої радіації, зміни температури, багато хімічних речовин (ліки, пестициди, деякі харчові добавки) і біологічні фактори (віруси).
Мутації можуть характеризуватися змінами на рівні всього хромосомного комплексу або на рівні окремої хромосоми (хромосомні перебудови). Хромосомні перебудови супроводжуються втратою, придбанням або зміною положення ділянки хромосоми, а також зміною числа хромосом. Захворювання, пов'язані зі зміною числа хромосом або хромосомними перебудовами у людини, носять назву хромосомних хвороб.
Інша група мутацій визначається змінами, що відбуваються на рівні гена (молекулярні зміни), і називаються генними мутаціями. Вони не виявляються цитогенетичних, а виявляються в зміні спадкових властивостей у нащадків. Генні мутації лежать в основі більшості спадкових захворювань.
§ 55. Спадкові хвороби, обумовлені генними мутаціями
Мутації гена виявляються в змінах структури молекул дезоксирибонуклеїнової кислоти (ДНК), т. Е. Призводять до зміни генетичного коду. В результаті порушується розвиток певних ознак і властивостей організму. Фенотипічно ці порушення можуть виявлятися на різних рівнях. Вони виявляються в деяких випадках на морфологічному рівні, і спадкові захворювання при цьому характеризуються перш за все явними аномаліями будови тіла і внутрішніх органів (полідактилія - багатопалості, синдактилія - зрощення пальців, мікроцефалія і ін.). В інших випадках на перший план виступають зміни властивостей організму на фізіологічному рівні (гіпертонія, дальтонізм, гемофілія та ін.). Нарешті, в особливу групу виділяють спадкові хвороби обміну речовин, при яких особливо чітко виявляються зміни на біохімічному рівні (фенілкетонурія, галактоземія та ін.).
Передача спадкових ознак, що виникли в результаті генних мутацій, може здійснюватися за домінантним або рецесивним типом, а також може бути зчепленої з підлогою.
Домінантним, або більш сильно котрі виявляють свою дію, може бути як нормальний, так і патологічний ген. Ген, властивості якого у потомства при цьому не виявляються або виявляються слабо, буде рецесивним по відношенню до домінантним.
§ 56. Хвороби, успадковані за домінантним типом
При домінантному типі успадкування один з батьків хворого також хворий. При великому числі дітей є співвідношення хворих і здорових, рівне 1: 1, тому що шанси отримати або не одержати мутантний ген приблизно однакові. Домінантне успадкування у людини може маскуватися явищем пенетрантности, т. Е. Невияв дії патологічного гена. Що володіють цим геном залишаються здоровими, але можуть передати дітям спадкове захворювання. Якщо в сім'ї є перший випадок домінантного захворювання (спорадичний), то мова йде про знову виникла мутації в зародковій клітці одного з батьків, який вважається здоровим.
За домінантним типом успадковуються такі ознаки як синдактилия (зрощення пальців рук), полідактилія (збільшення числа пальців до 6 або навіть 7), брахидактилия (вкорочення пальців на руках і ногах внаслідок недорозвинення середніх фаланг). До цієї ж групи спадкових патологій відноситься ахондроплазия, що характеризується карликовим ростом і непропорційним складанням з помітним укороченням рук і ніг внаслідок порушення росту епіфізарних хрящів довгих трубчастих кісток. Прогресуюча хорея (хорея Гентінгтона), що виявляється у віці 35-40 років і пізніше, характеризується тремтінням рук, мимовільними рухами (гіперкінез) і подальшим недоумством.
Як домінантна ознака передаються також множинний нейрофіброматоз (хвороба Реклінгаузена), вроджена атрофія слухового нерва, мозочкова атаксія і багато інших захворювань.
§ 57. Хвороби, успадковані за рецесивним типом
Ген, що визначає хвороба, не проявляється в присутності його алелі, який зумовлює нормальні функції організму. Приблизно половина спадкових хвороб людини обумовлена рецесивними мутаціями. Захворювання зазвичай виявляється у братів і сестер при зовні здорових батьків. При рецесивним успадкування може довго існувати приховане гетерозиготное носійство мутантного гена. Захворювання виникає при шлюбі двох гетерозигот - носіїв внаслідок утворення патологічної гомозиготи. У шлюбі гетерозигот хвора приблизно четверта частина дітей. Половина дітей є гетерозиготами. Родинні шлюби підвищують ймовірність виникнення рецесивних спадкових захворювань.
Таблиця 11. Спадкові хвороби обміну амінокислот, зумовлені недостатньою активністю або відсутністю ферментівАмінокислотаХворобаФермент
фенілаланін Фенилкетонурия
фенілпіровиноградна олігофренія
Фенілаланінгідроксилази Тирозин Тирозиноз оксидазой оксіфенілпіровіноградной кислоти Тирозин Алкаптонурия оксидазой гомогентизиновой кислоти Тирозин Альбінізм тирозинази Аргінін Аргінінсукцінатурія
ксантинурія
Аргин інсукціназа
ксантиноксидаза

До аномалій розвитку, успадковані за рецесивним типом, відносяться мікроцефалія (значне зменшення розмірів черепа і мозку за рахунок недорозвинення великих півкуль), іхтіоз (захворювання, при якому поверхня шкіри покрита великими кровоточать тріщинами і ороговевшими пластами, такі діти незабаром вмирають або народжуються мертвими). За рецесивним типом успадковуються також дефекти амінокислотного обміну (альбінізм, фенілкетонурія, алкаптонурія і ін.) (Табл. 11).
Порушення обміну фенілаланіну (фенілкетонурія). Причиною захворювання є нестача ферменту фенілаланінгідроксилази в печінці, внаслідок чого блоковано перетворення фенілаланіну в тирозин (рис. 6). Концентрація фенілаланіну в крові досягає 0,2-0,6 г / л (в нормі близько 0,015 г / л). Продукти його метаболізму, зокрема кетокислоту - фенілпіруват, надають токсичну дію на нервову систему. Нервові клітини кори головного мозку руйнуються і заміщуються мікрогліальними елементами. Розвивається фенилпировиноградная олігофренія.
Починаючи з 6-місячного віку, у дитини відзначається відставання розумового розвитку, знебарвлення шкіри і волосся, підвищення тонусу м'язів і основного обміну, епілепсія, мікроцефалія і ін.
Просвітлення кольору шкіри і волосся розвивається через недостатність вироблення меланіну, так як в результаті накопичення фенілаланіну блокується метаболізм тирозину.
Знижується синтез катехоламінів, знижується рівень інших вільних амінокислот в плазмі крові. Збільшується виділення кетонових тіл з сечею.
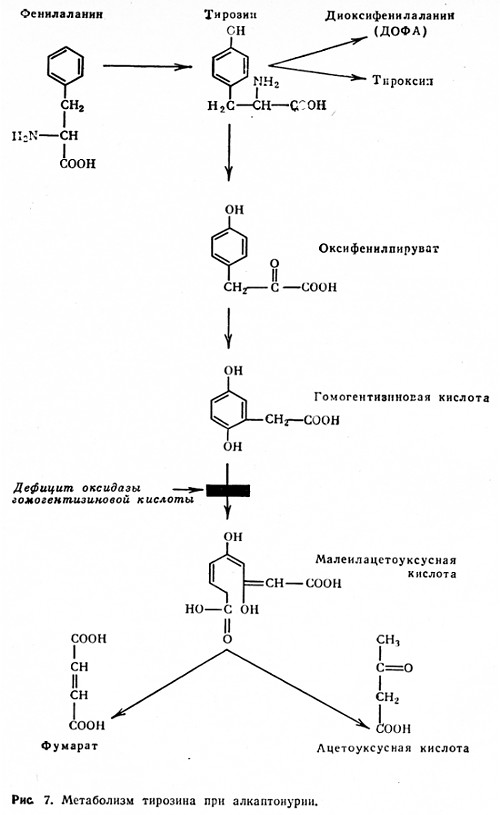
Порушення обміну гомогентизиновой кислоти (продукту метаболізму тирозину) - алкаптонурія - виникає при нестачі ферменту оксидази гомогентизиновой кислоти (рис. 7). При цьому гомогентізіновая кислота не переходить в малеілацетоуксусную кислоту (не відбувається розриву гидрохиноновую кільця). У нормальних умовах гомогентізіновая кислота в крові не визначається. При недостатності ферменту гомогентізіновая кислота з'являється в крові і виводиться з організму з сечею. Відзначається характерне потемніння сечі, особливо в лужному середовищі.
Відкладення похідних гомогентизиновой кислоти в тканинах викликає пігментацію сполучної тканини - охроноз. Пігмент відкладається в суглобових хрящах, в хрящах носа, вушних раковинах, в ендокардит, великих кровоносних судинах, нирках, легенях, в епідермісі. Алкаптонуріі часто супроводжує нирковокам'яна хвороба.
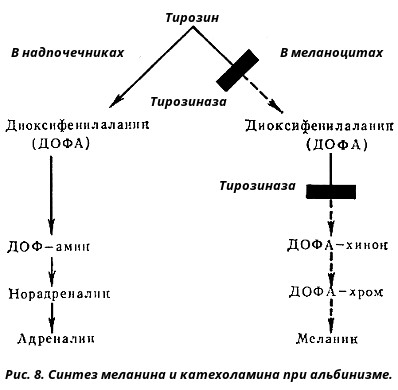
Порушення обміну тирозину - альбінізм. Причиною захворювання є нестача ферменту тирозинази в меланоцитах - клітинах, які синтезують пігмент меланін (рис. 8).
При відсутності меланіну шкіра набуває молочно-білий колір з білястим оволосением (альбінізм), спостерігаються світлобоязнь, просвічування райдужної оболонки, зниження гостроти зору. Сонячне опромінення викликає запальні зміни шкіри - еритема.
Альбінізм може супроводжуватися глухотою, німотою, епілепсію, полідактилія і олігофренію. Інтелект таких хворих чаші нормальний.
Порушення обміну гистидина. Мастоцитоз - спадкова хвороба, що супроводжується посиленою проліферацією гладких клітин. Причиною захворювання вважають підвищення активності гістідіндекарбоксілази - ферменту, що каталізує синтез гістаміну. Гістамін накопичується в печінці, селезінці та інших органах. Хвороба характеризується ураженням шкіри, порушеннями серцевої діяльності і функції шлунково-кишкового тракту. Відзначається підвищена екскреція з сечею гістаміну.
Цистиноз. Спостерігається при вродженому дефекті реабсорбції в канальцях нирок цистину, цистеїну та інших нециклічних амінокислот. Екскреція амінокислот з сечею може збільшуватися при цьому в 10 разів. Екскреція цистину і цистеїну зростає в 20-30 разів. Цистин відкладається в нирках, селезінці, шкірі, печінці. Цистиноз супроводжується глюкозурією, гіперкаліуріей, протеїнурією і полиурией.
При цістінуріі екскреція цистину може збільшуватися до 50 разів у порівнянні з нормою, супроводжуючись пригніченням реабсорбції лізину, аргініну і орнитина в ниркових канальцях. Рівень цистину в крові не перевищує норми. Не виявлено порушень в проміжному обміні цих амінокислот. Підвищена екскреція амінокислот може привести до порушень синтезу білка і білкової недостатності.
§ 58. Спадкові хвороби, зчеплені з підлогою
Деякі рецесивні спадкові захворювання успадковуються сцепленно з Х-хромосомою. Для Y-хромосоми ще не описано патологічних мутацій.
Так успадковується гемофілія А і гемофілія В. Гемофілія А (класична форма захворювання) залежить від нестачі антигемофильного глобуліну (фактора VIII), а гемофілія В обумовлена дефіцитом фактора Крістмаса (IX фактор). Захворювання практично зустрічається тільки у чоловіків з частотою приблизно 1:10 000 При шлюбі здорової, але гетерозиготною по гемофілії жінки зі здоровим чоловіком половина їхніх синів буде вражена на гемофілію. Дочки не захворіють, але половина з них (гетерозиготи) є кондукторами хвороби. Чоловіки, хворі на гемофілію, і здорові дружини мають здорових синів, але всі їхні дочки є кондукторами хвороби. Гемофілія у дівчаток зустрічається вкрай рідко, так як шлюби чоловіки, хворого на гемофілію, з жінкою-кондуктором рідкісні і дівчатка в таких шлюбах часто гинуть.
Дальтонізм або дальтонізм наслідується подібним же чином, сцепленно з Х-хромосомою. В даний час налічується понад 90 зчеплених з Х-хромосомою спадкових захворювань людини. Велике число з них пов'язано з різними аномаліями очей. До цієї групи належить вроджена куряча сліпота, що характеризується відсутністю сутінкового зору, пігментний ретиніт і ін.
§ 59. Хромосомні хвороби
Іншу форму патології в порівнянні з захворюваннями, викликаними генними мутаціями, представляють хромосомні хвороби, які, як правило, не передаються нащадкам і тому зустрічаються в сім'ї як спорадичні випадки. Виникнення хромосомних хвороб пов'язано з порушенням кількості або структури хромосом. При нерозходженні однієї з пар хромосом під час мейозу і попаданні цієї пари в яйцеклітину її ядро отримує від матері набір з додатковою хромосомою (рис. 9). Якщо ж неразошедшаяся пара потрапляє в Направітельний тільце, то в ядрі виникає нестача однієї з хромосом. При заплідненні такого яйця сперматозоїдом в зиготі замість нормального диплоїдного набору виникає порушений, так званий анеуплоїдних набір з нестачею або надлишком хромосом. Організм, який розвинувся з такою зиготи, буде містити змінений набір хромосом у всіх клітинах тіла.
Поєднання в зиготі XXZ- або XY-хромосом обумовлює нормально збалансований набір генів і детермінує нормальний розвиток жіночого і чоловічого плода. Відсутність Х-хромосоми в зиготі (YO) призводить до виникнення нежиттєздатного плода. Каріотипи XXY, ХО і XXX ведуть до виникнення життєздатних зародків, проте їх розвиток протікає ненормально. Так виникають хромосомні захворювання, які не мають сімейного характеру і зустрічаються частіше серед нащадків літніх матерів, у яких частіше буває нерасхождение хромосом в статевих клітинах. Для діагностики нерасхожденія хромосом досліджують хромосомний набір (каріотип) в клітинах тіла, наприклад в клітинах кісткового мозку, або визначають статевий хроматин, що зустрічається на периферії ядра і забарвлюється основними фарбами тільце, яке в нормі зустрічається лише в клітинах тіла жінок і відсутній у чоловіків (тільця Барра).
XXY-cиндром, або синдром Клайнфельтера. При цьому захворюванні, зустрічається у 1-2 чоловіків з 1000, в клітинах тіла міститься не 46, а 47 хромосом, причому є три статеві хромосоми XXY. Для чоловіків з синдромом Клайнфельтера характерні високий зріст, астенічний статура, довгі ноги, слабо розвинена мускулатура, зниження сперматогенезу, інертність і іноді розумова відсталість. У клітинах тіла виявляється статевий хроматин, так як є дві Х-хромосоми (хроматінположітельние чоловіки).
ХО-синдром, або синдром Шерешевського-Тернера. Даній синдром вінікає, коли зигота має лишь одну Х-хромосому при відсутності Y-хромосоми. Розвивається плід жіночої статі з каріотипом 45X0. Хворі характеризуються низькорослістю і відсутністю гонад. Статеві органи в інфантильному стані, типові первинна аменорея і безпліддя. Одним з необов'язкових ознак є наявність широкої шкірної складки на шиї від потилиці до надпліччя (крилоподібна складка). У клітинах слизової порожнини рота відсутній статевий хроматин, що дозволяє здійснювати ранню діагностику.
ХХХ-синдром, або синдром трисомії по Х-хромосомі. Вперше описаний у жінок з наявністю двох тілець статевого хроматину при каріотипі 47 XXX. У таких жінок іноді спостерігається гіпофункція яєчників і безпліддя, але в інших випадках статева функція нормальна і жінки можуть мати дітей.
Порушення хромосомного набору можуть стосуватися не тільки статевих хромосом, а й кожної з 22 пар аутосом. Так, у людини описані трисомії по 21 хромосомі, а також по хромосомах з груп 13-15, 17 і 18, 22.
Хвороба Дауна, або трисомія по 21 парі хромосом, є різновидом олігофренії. У соматичних клітинах хворих дітей є 47 хромосом замість 46. Характерно зменшення розмірів черепа, плоский потилицю, косе розташування очей, короткі кінцівки, відставання в рості. Перенісся уплощена, очі широко розставлені. Є всі переходи від помірної розумової відсталості до повної ідіотії. Відзначається специфічне зміна згинальних складок і шкірних візерунків на долонях, зокрема наявність однієї великої згинальній складки на долоні замість трьох.
Синдром трисомії по 13-15 парам хромосом (D-трисомія) характеризується рядом дефектів розвитку з боку очей і порожнини рота, відзначаються також полідактилія і дефекти з боку серця. Найбільш характерними рисами є м'язова гіпертонія і заяча губа.
Синдром трисомії по 17 і 18 парам хромосом (Е-трисомія) супроводжується множинними аномаліями розвитку. Характерні слабо розвинена нижня щелепа, дуже маленький рот, низьке стояння вух, брахі- і синдактилія, особливе становище вказівного пальця над 3-м пальцем, глибока дебільність.
Хромосомні хвороби можуть виникати в результаті внутріхромосомних або міжхромосомні перебудов (хромосомні аберації) і не завжди бувають пов'язані зі зміною загального числа хромосом. Абераціями хромосом називають зміни структури хромосом, при яких відбувається або порушення її безперервності, або перекомбінація ділянок хромосоми або декількох хромосом. Частина хромосоми може бути втрачена, що позначається як делеція. Довжина хромосоми може бути збільшена внаслідок повторення деякого сегмента - так звана Дуплікація. Транслокацией позначається перенесення частини хромосоми на іншу хромосому. Наприклад, деякі форми хвороби Дауна пов'язані з перенесенням частини 21 хромосоми на 15-ю пару хромосом (21/15). Нарешті, можлива інверсія, коли той чи інший сегмент хромосоми виявляється перевернутим.
Хромосомніаберації виникають під впливом різних факторів зовнішнього середовища (фізичних, хімічних, біологічних) і в даний час можуть бути виявлені цитогенетическими методами. Зокрема можна проводити облік хромосомних аберацій в кістковому мозку або лімфоцитах периферичної крові осіб, що контактують в тих чи інших умовах з мутагенну фактором. Хромосомніаберації, як правило, викликають загибель плода в ембріональній стадії або повне безпліддя. Тому хромосомніаберації, як і хромосомні хвороби, пов'язані зі зміною числа хромосом, не накопичуються з покоління в покоління, як при генні мутації, а виникають в кожному поколінні заново.
§ 60. Патогенез спадкових хвороб
Питання про механізм розвитку спадкових захворювань і, зокрема, про механізм дії мутантного гена є складним. Відповідно до теорії Бідла і Татум первинне дію гена полягає в тому, що кожен ген програмує синтез певного ферменту. Порушення в ферментної системі тягне за собою випадання біохімічної реакції і, як наслідок, порушується розвиток певних ознак організму. Таким чином, розвиток спадкових ознак відбувається за схемою: ген - фермент - біохімічна реакція - ознака. Є підстави припускати, що ген контролює синтез одного ферменту. Гіпотеза "один ген - один фермент" була висунута при вивченні метаболізму бактерій і грибів. В даний час ця гіпотеза кілька модифікована і відома як гіпотеза "один ген - один поліпептид".
З цих позицій в даний час пояснюється патогенез багатьох спадкових обмінних захворювань, зокрема фенілкетонурії. Дія гена фенілкетонурії визначає недостатність або відсутність одного ферменту - фенілаланінгідроксилази. Всі інші симптоми даного захворювання можна пояснити, виходячи з цього зміни (накопичення фенилпировиноградной кислоти, отруєння організму продуктами метаболізму, слабоумство).
Ген може діяти не на один, а на багато ознак, що позначається як плейотропізм. Так, при арахнодактіліі, або хвороби Марфана, основний синдром подовження пальців рук і ніг часто поєднується з іншими аномаліями скелета, зміщенням кришталика очей і вродженим пороком серця.
Більшість ознак залежить від спільної дії багатьох генів. Розвиток будь-якої морфологічної системи залежить від величезної кількості біохімічних реакцій і зміна в одній з них може змінити весь курс морфогенезу.
В даний час слід визнати, що всі спадкові дефекти в основі патогенезу мають молекулярні зміни в полінуклеотидних ланцюга ДНК і, отже, в структурі або інтенсивності синтезу окремих білків. Зміни білків на молекулярному рівні вивчені для гемоглобинопатий (наприклад, зміни в структурі гемоглобіну при серповидноклеточной анемії), для ряду ферментопатий.
Що стосується хромосомної патології, то слід мати на увазі, що при зміні числа хромосом або при хромосомних абераціях генний склад генотипу якісно не змінюється. При хромосомних захворюваннях змінюється лише "доза" окремих генів і кількісний генний і хромосомний баланс, отже, порушується збалансоване взаємодія генів в складі генотипу. При цьому, якщо хромосомні порушення сумісні з життям, виникають серйозні розлади онтогенетичного розвитку організму і патологічні зрушення в різних системах і морфологічних структурах.
§ 61. Поширеність спадкових хвороб і соціальні аспекти генетики людини
Соціальний і економічний прогрес суспільства змінює структуру захворювань людини. У XX столітті завдяки науково-технічному прогресу та успіхам розвитку медицини значно знизилася інфекційна захворюваність і знайдені засоби захисту проти багатьох екзогенних хвороботворних чинників. У зв'язку з цим питома вага спадкової патології має тенденцію до підвищення.
В даний час описано понад 1500 різних спадкових захворювань. Частота появи окремих спадкових хвороб, як правило, невелика; вона відповідає 1: 5000-1: 100 000 і рідше. Однак деякі спадкові захворювання зустрічаються частіше (шизофренія 1: 100, діабет 1: 1000). Аномалія колірного зору - дальтонізм - вражає 8% чоловіків і 0,5% жінок. За даними ВООЗ, близько 4% новонароджених страждає тими чи іншими генетично зумовленими дефектами. У країнах з розвиненим охороною здоров'я серед дітей, госпіталізованих в лікарні загального профілю, 20-25% складають діти зі спадковими захворюваннями.
Звідси зрозуміла важливість подальшого дослідження шляхів профілактики спадкових захворювань і своєчасного їх лікування. Виключне значення в соціальному плані має боротьба із захворюваннями, при Которн спадковість виступає як сприяючий чинник (гіпертонічна хвороба, ревматизм, подагра, алергічні захворювання). При цьому усунення зовнішніх агентів, що сприяють прояву хвороби, може повністю попередити розвиток хвороби.
Додатково: див. Медична генетика карти сайту "Акушерство"
продовження: Глава 2. Роль конституції в патології
до змісту

